Insights into the Structural and Energetic Descriptions of Ubiquitin Specific Protease 7 (USP7) Catalytic Mechanisms by Hybrid QM/MM Simulations
Abstract
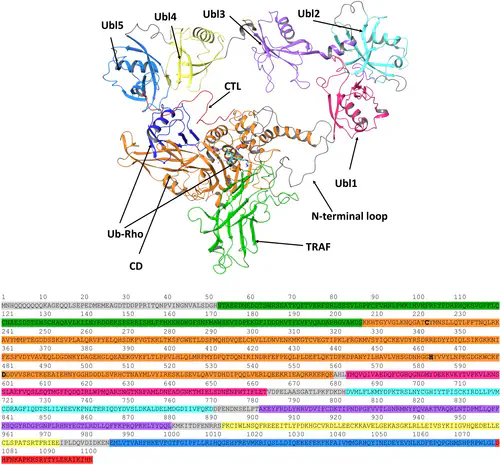
In this work, we conducted the first computational study of the USP7 reaction mechanism with the substrate Ubiquitin-Rhodamine 110-G (Ub-Rho) using a robust methodology that integrated homology modeling, classical molecular dynamics (MD) simulations, protein-protein interaction fingerprints (IFPs) analysis, principal component analysis (PCA), clustering, and hybrid quantum mechanics/molecular mechanics (QM/MM) simulations with the adaptive string method (ASM). Our results provide insights into the dynamic nature of USP7 enzyme-substrate complexes, offering a detailed structural description of the most relevant conformational changes observed in our simulations. These findings can serve as a reference for modeling more complex USP7 enzyme-substrate systems. Additionally, we characterized the protein-protein interactions of Ub-Rho residues at the P5 to P1’ positions with the catalytic domain’s active site and identified the geometries of stationary states along the minimum free energy path (MFEP). This information can be leveraged to design new potent and selective inhibitors targeting USP7 activity.