Computational Estimation of Residence Time on Roniciclib and Its Derivatives against CDK2: Extending the Use of Classical and Enhanced Molecular Dynamics Simulations
Abstract
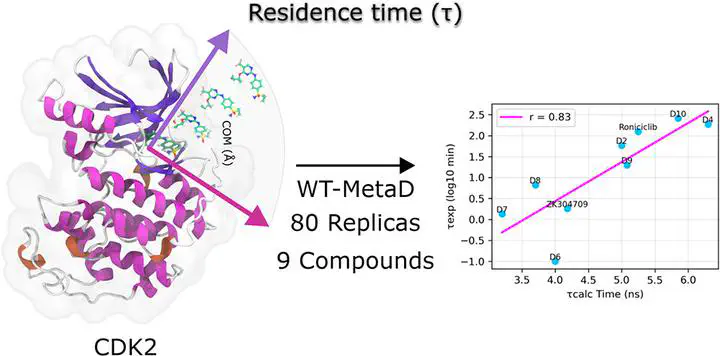
Residence time is a crucial parameter for assessing the functional efficacy of drugs, quantifying the duration of a drug’s binding to its target protein. It is directly related to therapeutic effects and the dosing regimen. Several factors can influence the residence time, including drug–protein binding kinetics and the unbinding pathways. Understanding the efficacy of a drug requires the characterization of both its binding kinetics and unbinding pathways from the drug–protein complex. By employing our previous computational protocol that uses enhanced sampling techniques such as well-tempered metadynamics (WT-MetaD) and classical molecular dynamics (cMD) simulations, it was possible to elucidate the inhibitor unbinding pathways and identify molecular determinants that extend the residence time in a set of cyclin-dependent kinase 2 (CDK2) inhibitors. In this study, using WT-MetaD, the relative residence times of roniciclib and eight derivatives were quantified on the nanosecond timescale. Notably, substituting the R5 position of the aminopyridine core with larger substituents significantly prolonged the computational residence time, which correlated well with experimental data (R2 = 0.83). Our computational simulations reveal the critical importance of specific amino acids, including Phe80, Lys33, and Asp145, in maintaining the stability of the protein–inhibitor complex. These residues are key in keeping the hydration network around them, affecting the inhibitor binding duration. The hydrogen bond interaction between residue Asp145 and roniciclib and its derivatives is particularly noteworthy, significantly influencing the electrostatic contribution to the binding free energy when the halogen substituent size increases. Furthermore, our analysis of protein flexibility at the C-terminus and N-terminus angles revealed a relationship with the size of the R5 substituent in the bound inhibitor, supported by principal component analysis. Additionally, different unbinding pathways were proposed, where it was found that inhibitors can dissociate from the CDK2 binding site through two principal routes: the α-helix D and β-1 and β-2 segments.