Understanding Substrate Binding and Reactivity of Stearoyl-CoA Desaturase (SCD1) through Classical and Multiscale Molecular Dynamics Simulations
Abstract
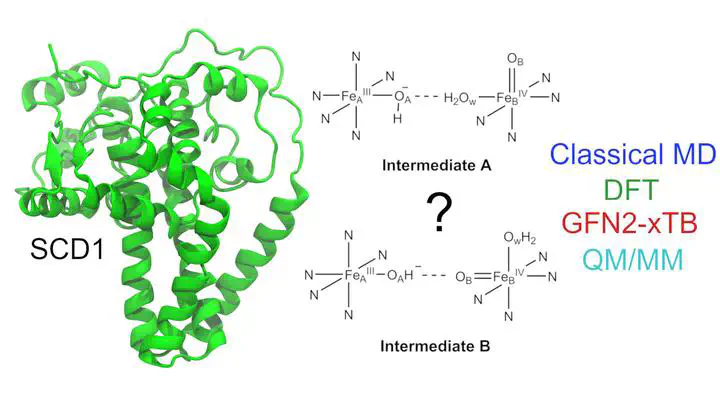
Stearoyl-CoA desaturase (SCD1) plays an important role in the metabolism of fatty acids and is a promising therapeutic target. However, the underlying mechanism of SCD1, as well as other transmembrane nonheme diiron enzymes, remains poorly understood. This study builds upon a previous density functional theory (DFT) cluster model study which proposed a potential reactive intermediate of SCD1. We assessed its dynamical properties by employing classical molecular dynamics (MD) simulations. The simulations revealed that the proposed intermediate lacks the ability to form a favorable reactive complex with stearoyl-CoA, highlighting the significance of a conserved asparagine residue in controlling the substrate�s orientation. Motivated by these observations, we proposed a modified intermediate in which a water molecule is strategically placed to stabilize the conserved asparagine residue. Subsequent classical MD simulations showed that the modified intermediate is able to form a reactive complex with the substrate, consistent with the experimentally observed selectivity of SCD1. A cluster model DFT study showed that the modified intermediate is of similar reactivity as the previously reported intermediate. The free energy barrier for the first hydrogen atom abstraction step by the modified intermediate was estimated to be accessible. The estimate is based on a hybrid quantum mechanics/molecular mechanics (QM/MM) approach utilizing the efficient semiempirical GFN2-xTB method. Considering its computational efficiency, GFN2-xTB seems to be a promising tool for the study of complex transition metal systems. Overall, our findings reveal important structure�function relationships in SCD1, uncovering an interplay between conserved residues and regioselectivity which advances our understanding of the entire class of transmembrane nonheme diiron enzymes.