Machine Learning Potentials for Chemical Reactions in Solution
In our research group, we develop machine learning potentials (MLPs) to perform molecular dynamics simulations with ab initio accuracy.
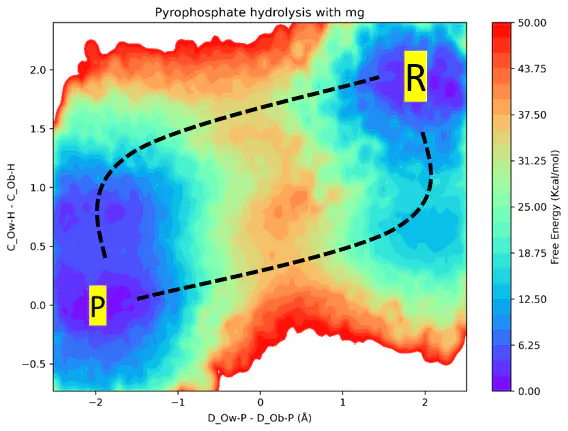
MLPs enable the study of chemical reactivity in solution with a radical reduction in computational cost compared to traditional ab initio methods, making large-scale simulations feasible. This enables significantly longer simulations and extensive sampling through enhanced techniques such as On-the-Fly Probability Enhanced Sampling (OPES) and Transition Path Sampling (TPS), facilitating the efficient exploration of reaction mechanisms. Currently, we are employing MLPs to investigate pyrophosphate hydrolysis and the catalytic role of cofactors and pH in this reaction. To train these models and generate comprehensive datasets, we employ the DeePMD-kit v2 software package integrated with the ArcaNN framework, designed for constructing training datasets for reactive MLPs. In the figure we show one of the free energy surfaces generated using OPES with a MLP.
This work is carried out in collaboration with Prof. Damien Laaage